Quantum mechanics
Quantum mechanics and its computational implementation is central to many aspects of modelling research at Manchester.
Atoms and molecules
Both ab initio (wave function) theory (WFT) and density functional theory (DFT) methods are employed to study electronic, geometric, energetic and magnetic properties and reactivity of molecules and solids from all over the periodic table.
- Quantum Chemical Topology (QCT) is developed and applied in the group of Prof Paul Popelier, at both interpretative and predictive levels, to intermolecular interactions, bioisosterism, chemical bonding, force field design, pKa prediction, molecular simulation, QSAR/QSPR and Raman Optical Activity (ROA) spectra.
- Radio- and heavy element chemistry is studied by both DFT and WFT, as well as hybrid methods such as QM/MM. Focus is on fundamental f element chemistry, as well as applied environmental projects in actinide transport and reactivity at mineral surfaces, and nuclear waste remediation and storage.
- Multiconfigurational WFT techniques are central to studying molecular magnetism, with focus on the prediction of EPR and NMR spectra of paramagnetic lanthanide and actinide molecules.
- DFT is applied to biologically relevant systems such as the oxygen evolving complex of Photosystem II, the manganese complex active site of catalase and semiquinone free radicals involved in crucial electron transfer reactions in photosynthesis (Dr Patrick O’Malley). Another focus is the role of quantum mechanics and fast dynamics in enzyme catalysis, underpinning new industrial biotechnology and synthetic biology projects.
Figures
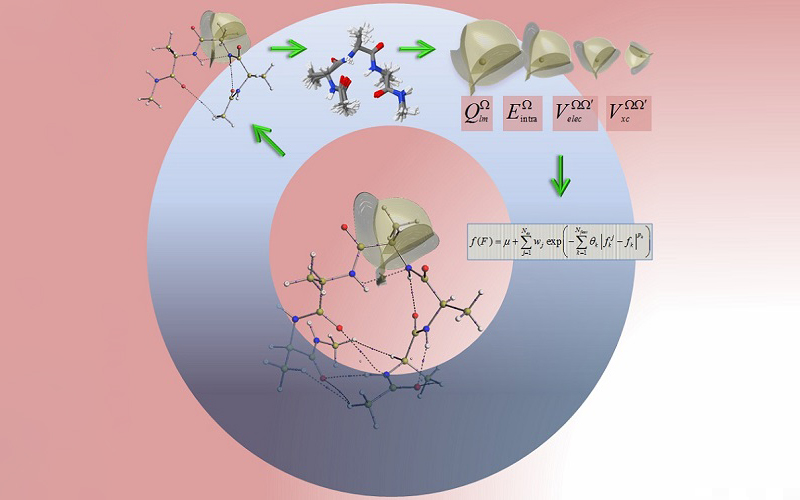
QCT provides topological atoms for the novel protein force field FFLUX, which uses the machine learning method kriging to model polarisation, charge transfer, bond strength, stereo-electronic energy and dispersion (click to enlarge).
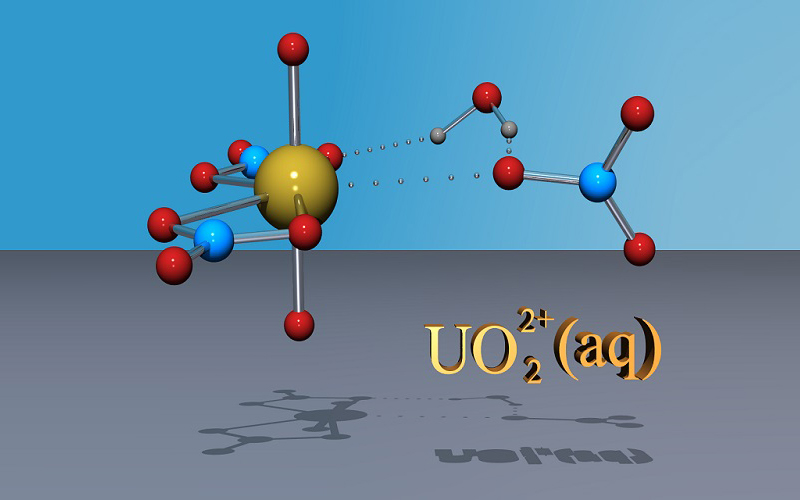
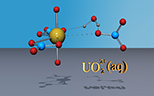
Simulation of the reactivity and sorption of uranyl at the aqueous interface of mineral surfaces (Dr Neil Burton).
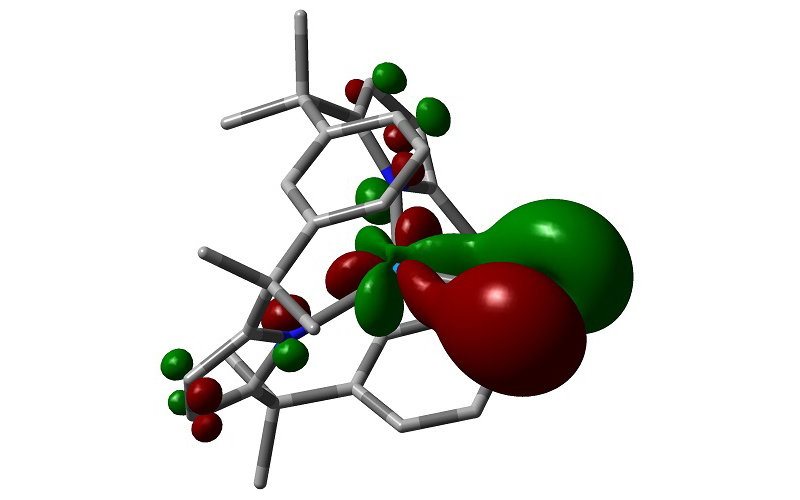
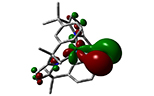
Neptunium-iodine bonding molecular orbital in [(LAr)NpI] (LAr = trans-calix[2]benzene[2]pyrrolide) (Prof Nik Kaltsoyannis)
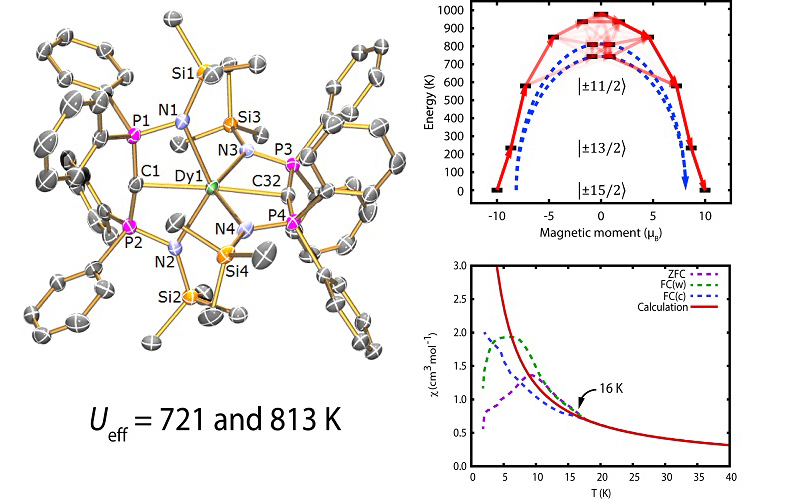
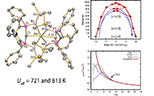
Designing coordination complexes to exhibit large magnetic anisotropy, resulting in intriguing magnetisation dynamics and magnetic memory effects (Dr Nick Chilton).
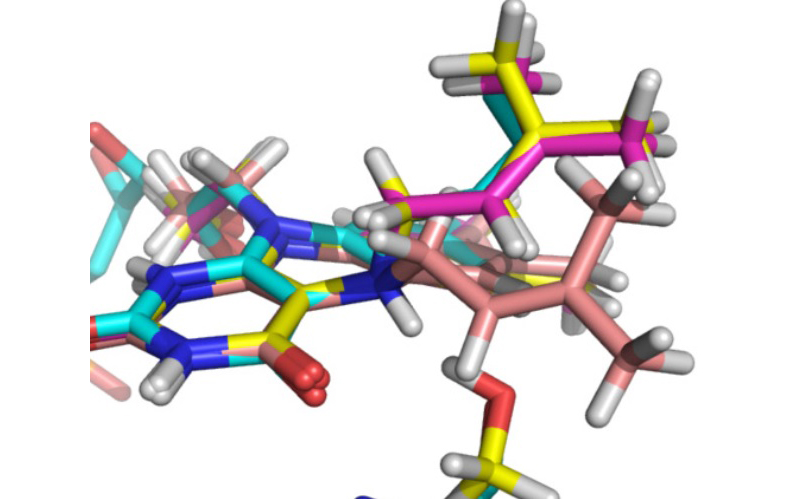
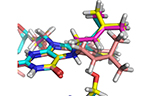
Overlay of DFT and X-ray crystal coordinates of a newly identified prenylated flavin cofactor (Dr Sam Hay).
Quantum mechanics – condensed matter
In this area, we note the research of the Condensed Matter Physics Group in the Department of Physics and Astronomy. Their research covers nanoscale and mesoscopic materials (graphene being a noteworthy example), metamaterials, and quantum fluids and solids. This group has strong links with the National Graphene Institute.